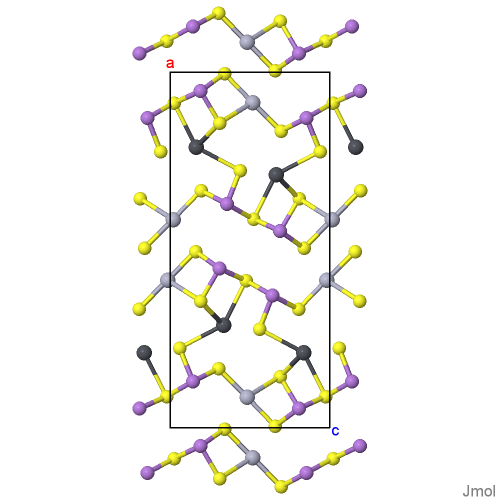
Daliranite, PbHgAs2S5: determination of the incommensurately modulated structure and revision of the chemical formula
Authors:Lanza, Arianna E.; Gemmi, Mauro; Bindi, Luca; Mugnaioli, Enrico; Paar, Werner H.
Journal:Acta Crystallographica, Section B 75 711-716 (2019)
DOI:https://doi.org/10.1107/S2052520619007340
B-IncStrDB ID: 15062EdDLkO Entry date: 2019-06-11 Last revision: 2021-12-12
average
Chemical data
Common Name: daliranite [ Help ]
Formula moiety: As2 Hg Pb S5 [ Help ]
Structural Formula Sum: As2 Hg Pb S5 [ Help ]
Formula weight: 717.9 Da [ Help ]
Crystallographic data and experimental details
Crystal system: orthorhombic [ Help ]
Space group name (H-M): Pnma [ Help ]
Space group nb.: 62 [ Help ]
Symmetry operations of the space group: (Show/hide table) [ Help ]
| Operation code | Operation in algebraic form |
|---|---|
| 1 | x, y, z |
| 2 | -x, -y, -z |
| 3 | -x+1/2, -y, z+1/2 |
| 4 | x+1/2, y, -z+1/2 |
| 5 | x+1/2, -y+1/2, -z+1/2 |
| 6 | -x+1/2, y+1/2, z+1/2 |
| 7 | -x, y+1/2, -z |
| 8 | x, -y+1/2, z |
a: 21.246(5) Å [ Help ]
b: 4.2897(9) Å [ Help ]
c: 9.5257(12) Å [ Help ]
α: 90 ° [ Help ]
β: 90 ° [ Help ]
γ: 90 ° [ Help ]
Volume: 868.2(3) Å3 [ Help ]
Z: 4 [ Help ]
Cell determination reflection Nb.: 2033 [ Help ]
θ(min) for cell determination: 0.05 ° [ Help ]
θ(max) for cell determination: 1.2 ° [ Help ]
Cell measurement temperature: 298 K [ Help ]
Absorption correction type: none [ Help ]
Experimental remarks: data have been collected by precession assisted 3D electron diffraction [ Help ]
Refinement details
Total nb. of reflections: 562 [ Help ]
Nb. of observed reflections: 401 [ Help ]
Intense reflections threshold: I>3σ(I) [ Help ]
Refinement based on: F [ Help ]
R(obs): 0.2435 [ Help ]
wR(obs): 0.2946 [ Help ]
R(all): 0.3222 [ Help ]
wR(all): 0.3025 [ Help ]
S(all): 13.93 [ Help ]
S(obs): 16.27 [ Help ]
Nb. of reflections: 562 [ Help ]
Nb. of parameters: 34 [ Help ]
Weighting scheme: sigma [ Help ]
Weighting scheme remarks: w=1/(σ2(F)+0.0001F2) [ Help ]
Δ/σ(max): 0.0002 [ Help ]
Δ/σ(mean): 0.0001 [ Help ]
Δρ(max): 0.45 e_Å-3 [ Help ]
Δρ(min): -0.40 e_Å-3 [ Help ]
Structural Information
Average Structure: (Show/hide table) [ Help ]
| Atom site label | Atom symbol | x | y | z | ADP type | Uiso/equiv | Symmetry multiplicity | Occupancy | Coords from (d)iffraction or (c)alculated | Coords restraints or constraints | Disordered cluster | Disordered group |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pb1 | Pb | 0.2886(7) | 0.25 | 1.3368(8) | Uani | 0.083(6) | 4 | 1 | d | ? | ? | ? |
| Hg1 | Hg | 0.4150(6) | 0.25 | 0.9831(7) | Uani | 0.065(5) | 4 | 1 | d | ? | ? | ? |
| As1 | As | 0.1286(13) | 0.356(3) | 1.1428(18) | Uiso | 0.030(6) | 8 | 0.5 | d | ? | ? | ? |
| As2 | As | 0.0526(12) | 0.356(3) | 0.8089(16) | Uiso | 0.019(6) | 8 | 0.5 | d | ? | ? | ? |
| S1 | S | 0.2233(15) | 0.25 | 1.060(2) | Uiso | 0.034(7) | 4 | 1 | d | ? | ? | ? |
| S2 | S | 0.1437(15) | 0.25 | 0.690(2) | Uiso | 0.036(7) | 4 | 1 | d | ? | ? | ? |
| S3 | S | 0.3337(16) | 0.25 | 0.805(2) | Uiso | 0.042(7) | 4 | 1 | d | ? | ? | ? |
| S4 | S | 0.4132(13) | 0.25 | 1.4762(19) | Uiso | 0.019(6) | 4 | 1 | d | ? | ? | ? |
| S5 | S | 0.4954(15) | 0.25 | 1.160(2) | Uiso | 0.040(7) | 4 | 1 | d | ? | ? | ? |
ADP components: (Show/hide table) [ Help ]
| Atom site label | Atom site symbol | U11 | U22 | U33 | U12 | U13 | U23 |
|---|---|---|---|---|---|---|---|
| Pb1 | Pb | 0.137(16) | 0.101(8) | 0.011(5) | 0 | -0.002332 | 0 |
| Hg1 | Hg | 0.118(14) | 0.069(6) | 0.009(5) | 0 | 0.001652 | 0 |
modulated
Chemical data
Formula moiety: As2 Hg Pb S5 [ Help ]
Structural Formula Sum: As2 Hg Pb S5 [ Help ]
Formula weight: 717.9 Da [ Help ]
Crystallographic data and experimental details
Crystal system: orthorhombic [ Help ]
Space group name (H-M): Pnma [ Help ]
Superspace group name: Pnma(00γ)0s0 [ Help ]
Symmetry operations of the superspace group: (Show/hide table) [ Help ]
| Operation code | Operation in algebraic form |
|---|---|
| 1 | x1,x2,x3,x4 |
| 2 | -x1+1/2,-x2,x3+1/2,x4+1/2 |
| 3 | -x1,x2+1/2,-x3,-x4+1/2 |
| 4 | x1+1/2,-x2+1/2,-x3+1/2,-x4 |
| 5 | -x1,-x2,-x3,-x4 |
| 6 | x1+1/2,x2,-x3+1/2,-x4+1/2 |
| 7 | x1,-x2+1/2,x3,x4+1/2 |
| 8 | -x1+1/2,x2+1/2,x3+1/2,x4 |
a: 21.246(5) Å [ Help ]
b: 4.2897(9) Å [ Help ]
c: 9.5257(12) Å [ Help ]
α: 90 ° [ Help ]
β: 90 ° [ Help ]
γ: 90 ° [ Help ]
Volume: 868.2(3) Å3 [ Help ]
Modulation dimension: 1 [ Help ]
Measured independent wave vectors: (Show/hide table) [ Help ]
| Wave vector id | q_x | q_y | q_z |
|---|---|---|---|
| 1 | 0.000000 | 0.000000 | 0.262(2) |
Z: 4 [ Help ]
Cell determination reflection Nb.: 10451 [ Help ]
θ(min) for cell determination: 0.05 ° [ Help ]
θ(max) for cell determination: 1.2 ° [ Help ]
Cell measurement temperature: 298 K [ Help ]
Absorption correction type: none [ Help ]
Experimental remarks: data have been collected by precession assisted 3D electron diffraction [ Help ]
Refinement details
Total nb. of reflections: 1503 [ Help ]
Nb. of observed reflections: 981 [ Help ]
Intense reflections threshold: I>3σ(I) [ Help ]
Refinement based on: F [ Help ]
R(obs): 0.2715 [ Help ]
wR(obs): 0.2989 [ Help ]
R(all): 0.3818 [ Help ]
wR(all): 0.3097 [ Help ]
S(all): 10.70 [ Help ]
S(obs): 12.87 [ Help ]
Nb. of reflections: 1503 [ Help ]
Nb. of parameters: 38 [ Help ]
Weighting scheme: sigma [ Help ]
Weighting scheme remarks: w=1/(σ2(F)+0.0001F2) [ Help ]
Δ/σ(max): 0.0009 [ Help ]
Δ/σ(mean): 0.0003 [ Help ]
Δρ(max): 0.45 e_Å-3 [ Help ]
Δρ(min): -0.40 e_Å-3 [ Help ]
Extinction method: none [ Help ]
Structural Information
Average Structure: (Show/hide table) [ Help ]
| Atom site label | Atom symbol | x | y | z | ADP type | Uiso/equiv | Symmetry multiplicity | Occupancy | Coords from (d)iffraction or (c)alculated | Coords restraints or constraints | Disordered cluster | Disordered group |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pb1 | Pb | 0.2885(5) | 0.25 | 1.3367(5) | Uani | 0.061(4) | 4 | 1 | d | ? | ? | ? |
| Hg1 | Hg | 0.4149(5) | 0.25 | 0.9835(5) | Uani | 0.063(4) | 4 | 1 | d | ? | ? | ? |
| As1 | As | 0.1285(8) | 0.371(2) | 1.1430(11) | Uiso | 0.031(3) | 8 | 0.5 | d | ? | ? | ? |
| As2 | As | 0.0517(7) | 0.365(2) | 0.8082(10) | Uiso | 0.027(3) | 8 | 0.5 | d | ? | ? | ? |
| S1 | S | 0.2236(10) | 0.25 | 1.0596(15) | Uiso | 0.027(4) | 4 | 1 | d | ? | ? | ? |
| S2 | S | 0.1430(9) | 0.25 | 0.6898(14) | Uiso | 0.019(4) | 4 | 1 | d | ? | ? | ? |
| S3 | S | 0.3344(11) | 0.25 | 0.8066(15) | Uiso | 0.026(4) | 4 | 1 | d | ? | ? | ? |
| S4 | S | 0.4134(8) | 0.25 | 1.4756(12) | Uiso | 0.007(4) | 4 | 1 | d | ? | ? | ? |
| S5 | S | 0.4962(10) | 0.25 | 1.1610(15) | Uiso | 0.030(5) | 4 | 1 | d | ? | ? | ? |
ADP components: (Show/hide table) [ Help ]
| Atom site label | Atom site symbol | U11 | U22 | U33 | U12 | U13 | U23 |
|---|---|---|---|---|---|---|---|
| Pb1 | Pb | 0.132(11) | 0.033(4) | 0.018(3) | 0 | -0.003(4) | 0 |
| Hg1 | Hg | 0.122(10) | 0.060(4) | 0.006(3) | 0 | 0.002(3) | 0 |
Fourier Wave Vectors (explicit: q_x,q_y,q_z or coefficients: q_1,q_2,...): (Show/hide table) [ Help ]
| Wave vector code | q_1 |
|---|---|
| 1 | 1 |
Occupation crenel coefficients: (Show/hide table) [ Help ]
| Atom site label | Center (x0) | Width |
|---|---|---|
| As1 | 0.390(5) | 0.5 |
| As2 | 0.314(5) | 0.5 |
Definition of the displacive (translational) Fourier series: (Show/hide table) [ Help ]
| Modulation code | Atom site label | Displacement axis | Wave vector code |
|---|---|---|---|
| Pb1x1 | Pb1 | x | 1 |
| Pb1y1 | Pb1 | y | 1 |
| Pb1z1 | Pb1 | z | 1 |
| Hg1x1 | Hg1 | x | 1 |
| Hg1y1 | Hg1 | y | 1 |
| Hg1z1 | Hg1 | z | 1 |
| S1x1 | S1 | x | 1 |
| S1y1 | S1 | y | 1 |
| S1z1 | S1 | z | 1 |
| S2x1 | S2 | x | 1 |
| S2y1 | S2 | y | 1 |
| S2z1 | S2 | z | 1 |
| S3x1 | S3 | x | 1 |
| S3y1 | S3 | y | 1 |
| S3z1 | S3 | z | 1 |
| S4x1 | S4 | x | 1 |
| S4y1 | S4 | y | 1 |
| S4z1 | S4 | z | 1 |
| S5x1 | S5 | x | 1 |
| S5y1 | S5 | y | 1 |
| S5z1 | S5 | z | 1 |
Displacive (translational) Fourier coefficients: (Show/hide table) [ Help ]
| Modulation code | Cosine coefficient | Sine coefficient |
|---|---|---|
| Pb1x1 | 0 | 0 |
| Pb1y1 | -0.0284 | 0.0865 |
| Pb1z1 | 0 | 0 |
| Hg1x1 | 0 | 0 |
| Hg1y1 | 0.0393 | 0.0529 |
| Hg1z1 | 0 | 0 |
| S1x1 | 0 | 0 |
| S1y1 | -0.0211 | 0.0555 |
| S1z1 | 0 | 0 |
| S2x1 | 0 | 0 |
| S2y1 | 0.0111 | 0.0498 |
| S2z1 | 0 | 0 |
| S3x1 | 0 | 0 |
| S3y1 | 0.0547 | 0.0185 |
| S3z1 | 0 | 0 |
| S4x1 | 0 | 0 |
| S4y1 | 0.0147 | 0.0209 |
| S4z1 | 0 | 0 |
| S5x1 | 0 | 0 |
| S5y1 | 0.0279 | -0.0315 |
| S5z1 | 0 | 0 |
Definition of the ADP Fourier series: (Show/hide table) [ Help ]
| Modulation code | Atom site label | Tensor element | Wave vector code |
|---|---|---|---|
| Pb1U111 | Pb1 | U11 | 1 |
| Pb1U221 | Pb1 | U22 | 1 |
| Pb1U331 | Pb1 | U33 | 1 |
| Pb1U121 | Pb1 | U12 | 1 |
| Pb1U131 | Pb1 | U13 | 1 |
| Pb1U231 | Pb1 | U23 | 1 |
| Hg1U111 | Hg1 | U11 | 1 |
| Hg1U221 | Hg1 | U22 | 1 |
| Hg1U331 | Hg1 | U33 | 1 |
| Hg1U121 | Hg1 | U12 | 1 |
| Hg1U131 | Hg1 | U13 | 1 |
| Hg1U231 | Hg1 | U23 | 1 |
ADP Fourier coefficients: (Show/hide table) [ Help ]
| Modulation code | Cosine coefficient | Sine coefficient |
|---|---|---|
| Pb1U111 | 0 | 0 |
| Pb1U221 | 0 | 0 |
| Pb1U331 | 0 | 0 |
| Pb1U121 | 0.015246 | 0.011714 |
| Pb1U131 | 0 | 0 |
| Pb1U231 | 0 | 0 |
| Hg1U111 | 0 | 0 |
| Hg1U221 | 0 | 0 |
| Hg1U331 | 0 | 0 |
| Hg1U121 | 0 | -0.008135 |
| Hg1U131 | 0 | 0 |
| Hg1U231 | 0 | 0 |